
單位簡介
分子與基因醫學研究組於民國85年元月獲准設立,隨即展開本組研究方向之規劃、實驗室之設立與研究人才延攬作業,歷經一年的積極推動,於民國86年七月分子與基因醫學研究組正式成立全面運作。並於民國100年5月獲董事會同意改制為分子與基因醫學研究所。
除院內計畫中的研究工作外,本所與中央研究院、台灣大學、陽明大學、清華大學、成功大學、榮民總醫院及其他研究單位都有相當密切的聯繫,並共同執行研究計劃。期以研究團隊的結合及資源的交流,引進並建立當前最新的遺傳及分子生物分析技術,應用於研究台灣常見疾病及癌症的分子機轉,研發出更有效的診斷與治療方法,以因應下一世紀醫學研究的挑戰。
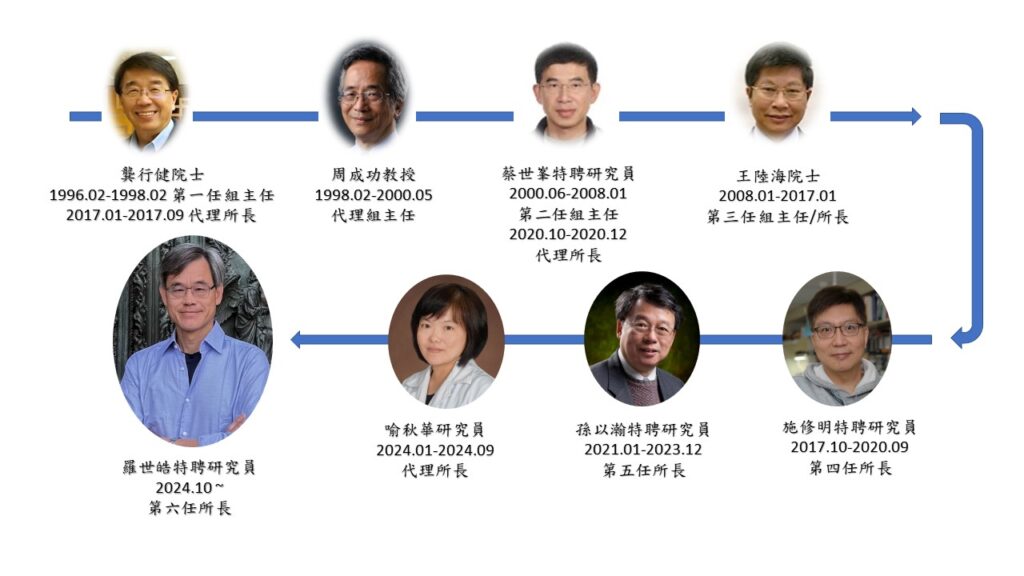
歷任組主任/所長
研究宗旨
-
- 發展及應用現代分子生物學及基因體技術平台,瞭解、預防及治療人類疾病的生醫研究。
-
- 促進台灣基因體學以及現代分子和遺傳學領域研究,以及相關生物科技產業之開發。
研究目標
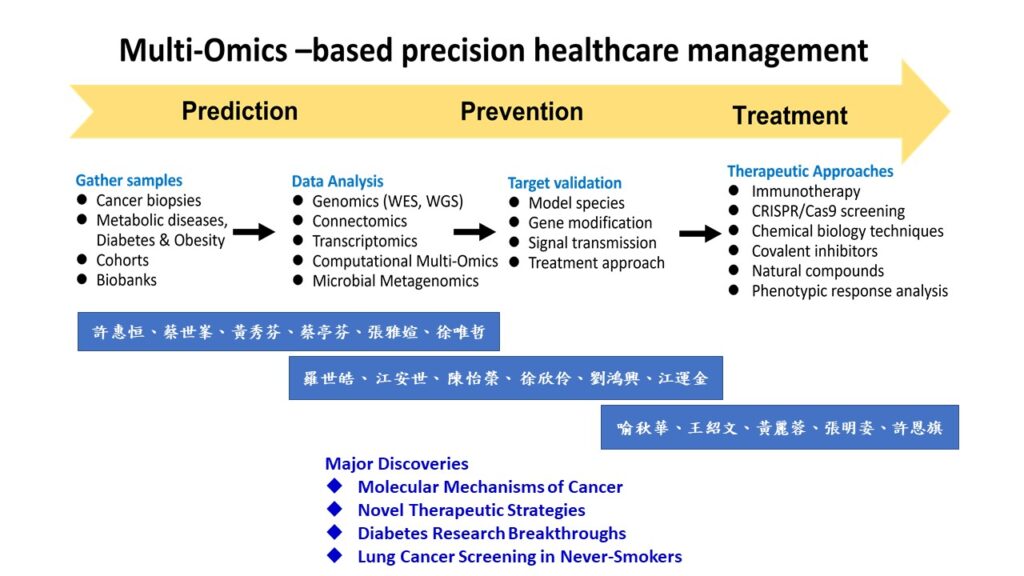
我們的研究專注於「以多體學為基礎之精準醫療管理——從預測到預防再到治療」。本所致力於利用多體學方法推動醫療進步,這種方法能透過預測、預防和治療各種疾病來實現精準醫療管理。
我們的合作框架整合了每位研究人員的多樣專業知識,通過多體學方法推動精準醫療的進展。大家合作研究主要聚焦於以下幾個項目:
-
- 預測:
-
- 利用基因體學、蛋白質體學和計算分析來識別疾病標誌並預測疾病的發生。
-
- 開發先進的成像技術以繪製神經網絡並理解疾病機制。
-
- 預防:
-
- 實施針對代謝疾病、癌症和神經系統疾病的精準預防策略。
-
- 利用生物資料庫和多體學數據開發早期干預方法。
-
- 治療:
-
- 發現並驗證新的藥物靶點和治療策略。
-
- 利用斑馬魚模型和其他動物模型進行新療法的臨床前測試。
-
- 開發針對各種疾病的免疫療法和個性化醫療方法。
-
- 2001 進階生物科技股份有限公司 (Functional Module Array and Novel Two-Hybrid System for Protein Linkage Maps) (2004終止)
-
- 2001 大展生命科技股份有限公司 (Recombinant baculovirus 在動植物體內蛋白質表現) (2014終止)
-
- 2001 國兆生物科技股份有限公司 (肝癌新穎基因研究計畫)
-
- 2011 伯森生物科技股份有限公司 (高通量細胞表現型篩選平台) (2014終止)
-
- 2012 豐技生物科技股份有限公司 ( TP53癌症基因突變篩選方法與相關資料庫)
-
- 2013 明欣生物科技股份有限公司 (多工細菌基因體定序及抗藥基因篩選平台)
-
- 2013 基龍米克斯生物科技股份有限公司 (多工細菌基因體定序及抗藥基因篩選平台)
-
- 2013 源資國際生物科技股份有限公司 (多工細菌基因體定序及抗藥基因篩選平台)
-
- 2019 財團法人國家實驗研究院 (原發性該細胞癌小鼠模式)
-
- 2021 台基盟生技股份有限公司 (精準醫療平台技術開發及產業應用)
-
- 2024 博蔚醫藥生物科技股份有限公司 (有效消除脂肪的方法及其相關措施)
-
- 2025 科進製藥科技股份有限公司 (基因轉殖魚肝癌模型)
(二) 研究成果
Part 1.
利用多基因風險分數揭示影響Metformin療效的糖尿病遺傳異質性
在精準醫療時代,個人化糖尿病治療策略的制定需將遺傳背景納入考慮。許惠恒特聘研究員兼副院長所領導的研究團隊,運用台灣精準醫療計畫(TPMI)所建置的大型基因與臨床資料庫,針對第二型糖尿病患者在台中榮總接受Metformin單一藥物治療的療效差異進行系統性分析。
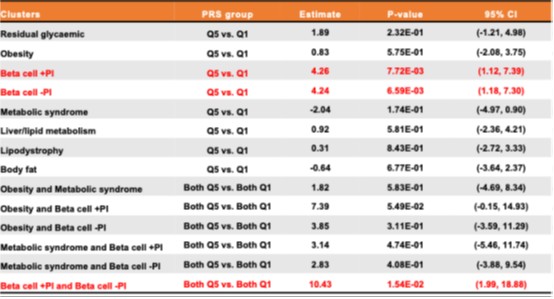
本研究針對2,507位第二型糖尿病患者,評估其在接受Metformin單一藥物治療半年後的空腹血糖與HbA1c變化,並結合近年大型GWAS研究所建立的8大SNP功能群,計算Partitioned-specific polygenic risk scores (pPRS)。研究發現,在β細胞功能異常相關風險群中(包含與proinsulin呈正相關與負相關兩群),pPRS最低20%(Q1)的患者,其Metformin治療後的空腹血糖下降幅度與pPRS最高20%(Q5)者,降幅4.3 mg/dL與4.4 mg/dL,合併分析更高達近10.7 mg/dL的差異(p<0.002,經Bonferroni校正後仍具顯著性)。此外,在HbA1c改善方面,於「β細胞功能異常且與proinsulin呈負相關」風險群中,pPRS最低者與pPRS最高者,降幅差異達0.18%(p=0.002)。
這些結果指出,即使在臨床起始數值、用藥劑量與基線代謝特徵相似的情況下,基因風險背景仍能顯著影響降血糖藥物治療反應。特別值得注意的是,β細胞功能異常這兩類群組的基因訊號,主要表現於胰島β細胞的染色質開放區域與神經內分泌細胞中,與Metformin已知之作用機制可能有所重疊,顯示這些遺傳差異可能透過影響胰島素分泌機制與GLP-1路徑調控,進而改變療效。此研究不僅為亞洲族群建立針對Metformin療效預測的遺傳學模型,也為糖尿病藥物反應個體化治療鋪路。
比較「β細胞功能異常且與正向proinsulin相關」與「β細胞功能異常且與負向proinsulin相關」這兩個群組中,polygenic risk score(多基因風險分數)最低20%(Q1)與最高20%(Q5)之間對使用metformin單一藥物治療半年的空腹血糖降幅反應差異
Part 2.
從臨床到基因:台灣MODY個案的整合式研究分析
MODY(Maturity-Onset Diabetes of the Young,年輕早發成年型糖尿病)是一種單基因遺傳性糖尿病,通常於25歲前發病,具有家族性遺傳模式,與典型的第一型或第二型糖尿病不同。MODY的患者胰島素分泌功能異常是其主要致病機制。不同的致病基因變異會導致臨床表現與治療反應有所差異,部分MODY亞型可用口服藥物治療、甚至無須使用胰島素,因此正確鑑別與基因診斷對於病患的個別化治療與家族成員的預防策略至關重要。許惠恒特聘研究員團隊與台中榮民總醫院新陳代謝科沈宜靜醫師、醫學研究部蕭自宏研究員合作,有如下成果:
一、發掘台灣年輕型糖尿病族群中的MODY致病變異與臨床意義
進行台灣首個針對MODY之前瞻性臨床與基因研究,收案對象包含三組:具典型臨床表現之疑似MODY個案(C-MODY),發病年輕但不符合MODY臨床特徵者(C-YOD),以及其家族成員。研究已完成80位受試者之全外顯子定序(WES),其中C-MODY佔58人。初步發現指出,有26.3%的受試者帶有MODY相關變異,以GCK、PAX4、WFS1、ABCC8與HNF1B為主。進一步分析顯示,13.8%帶有已知致病或可能致病等級的變異,全部來自C-MODY組,並首次在台灣族群中發現5.0%具雙基因(digenic)變異者,顯示這類基因交互作用對年輕型糖尿病臨床表現具有潛在影響力。
研究並發現,從無變異者、單一變異者至雙變異者之間,臨床參數如體重、BMI、腰圍、血壓、C-peptide、三酸甘油脂、GPT與尿中白蛋白皆呈漸進性下降趨勢。此發現不僅擴展對亞洲族群MODY基因圖譜的理解,也為未來精準分類與個別化照護奠定基礎。此研究結果在2025年美國糖尿病學會年會接受發表海報摘要。
二、MODY罕見變異在第二型糖尿病族群中的潛在貢獻:來自大型WES與TPMI資料的回溯性分析
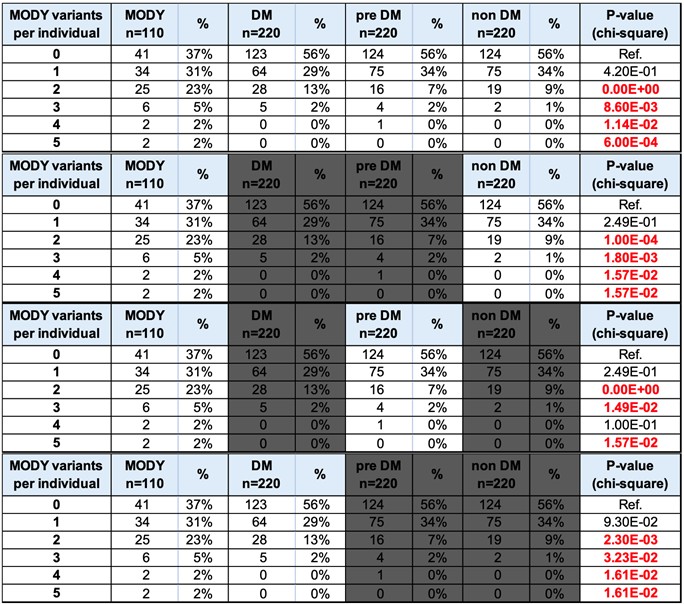
本研究團隊整合台中榮總與台灣精準醫療計畫(TPMI)之基因資料,利用70,000筆基因晶片與超過35,000筆全外顯子定序資料,進行針對第二型糖尿病患者之MODY罕見變異攜帶率分析。比較110名MODY患者與220名年齡、性別配對的糖尿病與非糖尿病對照組,結果顯示:MODY患者中具有兩個以上MODY基因變異者占32%,遠高於糖尿病患者(15%)與非糖尿病族群(10%)。這項結果不僅強化了MODY相關變異在台灣糖尿病族群中的潛在負荷,也指出傳統第二型糖尿病分類中可能隱含具單基因或雙基因致病潛力的個案,值得臨床加以篩檢與再分類。
此研究是亞洲首度於大規模糖尿病族群中系統性探討MODY罕見變異頻率與分布,並結合TPMI資源實證其在實際族群中所佔比例,為未來MODY臨床篩檢指標、健康照護政策與新型預防策略提供強有力的科學依據。
WES與TPMI資料揭示MODY變異在人群中的比例
Part 3.
揭示晚睡基因型與代謝症候群的因果關聯:跨族群的大型遺傳流行病學研究
人體晝夜節律與代謝健康息息相關,但其遺傳根源與代謝疾病風險的關聯仍未明朗。許惠恒特聘研究員團隊結合台灣人體生物資料庫(Taiwan Biobank, TWB)與英國生物資料庫(UK Biobank, UKB)超過30萬名參與者的資料,分析晚睡型(late chronotype)與代謝症候群之關聯。
結果發現,在雙族群中,晚睡者罹患代謝症候群的風險明顯較高,OR分別為1.17(TWB)與1.15(UKB),且皆通過Bonferroni多重校正標準(p值皆<1E-8)。進一步分析顯示,晚睡型與中央肥胖、高血糖、高三酸甘油脂密切相關,且PR值愈高者,其代謝異常風險顯著升高。以PRS最上10%之晚睡基因型個體相比於PRS最低10%且具早睡型者,其罹患代謝症候群的風險高出24%(TWB,OR=1.24, p=4.7E-5)。
尤具突破性的是,研究中首次在東亞族群辨識出與晚睡型顯著相關之全基因關聯SNP(rs9478358)及具橫跨族群訊號的新穎SNP(rs148523072),顯示某些基因調控機制對晝夜節律具有跨族群一致的影響力。利用一樣的PRS作為工具變項進行孟德爾隨機化分析後,在兩個族群中皆證實晚睡基因型與代謝症候群之間具有潛在的因果關聯,強化了其作為代謝疾病干預標的的可能性。此外,研究結果亦指出,即便個體具有高遺傳風險,但若實際生活中養成早睡習慣,其罹患代謝症候群風險仍可降低,顯示基因與行為之間存在交互作用。這項成果突顯晝夜節律與代謝功能之密切關聯,為未來睡眠行為調整介入、個人化預防策略與政策規劃提供科學依據。
Part 4.
建立兼具年齡與性別特異性的代謝症候群風險評分系統,打造糖尿病風險預測新典範
現行代謝症候群(Metabolic Syndrome, MetS)診斷標準雖已納入性別別腰圍與HDL膽固醇等門檻值,卻未能系統性考量年齡與性別在人群代謝異質性中的交互作用,導致風險辨識精度有限。為解決此盲點,許惠恒特聘研究員與徐唯哲專案技術助研究員及陳怡榮研究員合作,運用台灣生物資料庫近19萬筆年齡介於20歲以上個體的完整代謝數據,發展出一套結合年齡與性別加權模型的代謝症候群Z分數(MetS-Z score),並藉此精準預測第二型糖尿病(T2D)發生風險。
此研究透過主成分分析(PCA)依據年齡與性別,為五項MetS核心指標(腰圍、空腹血糖、收縮壓、三酸甘油脂、HDL)賦予差異化權重,再合成單一連續變數。在超過4萬名無糖尿病個體之縱向追蹤中,MetS-Z表現出卓越的預測力,五年內T2D發生風險預測AUC達男性0.76、女性0.80,顯著優於傳統ATP III診斷準則(P<0.0001),且橫跨不同年齡層均具穩定效能。值得一提的是,該分數與糖尿病發生呈高度線性關聯,每增加1分,T2D風險增加14.7倍(OR=14.7, 95% CI: 14.19–15.24),並可透過自動化平台(MetS-Z線上工具)進行個人化風險試算與風險分級。
此外,MetS-Z亦具辨識心血管疾病(CVD)潛力,為跨領域風險整合評估提供基礎。此項研究充分展現如何運用資料驅動模型與族群分層策略,強化傳統風險指標的解釋力與臨床應用性,不僅對台灣族群具高度代表性,亦為全球精準代謝醫學提供風險預測模式。此研究結果發表在2025年Journal of Diabetes Investigation。
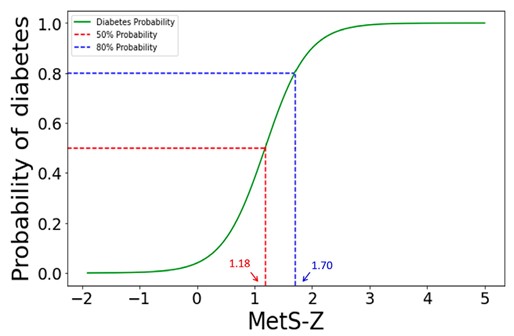
MetS-Z分數與第二型糖尿病風險之關聯曲線圖
本圖顯示個體之MetS-Z分數與罹患第二型糖尿病的累積機率之關係。研究發現,MetS-Z分數與糖尿病風險呈現高度正相關;當MetS-Z分數達到1.18時,糖尿病風險約為50%;而分數上升至1.70時,風險超過80%。每增加1分,發生糖尿病的機率將提高約14.7倍(95%信賴區間:14.19–15.24),凸顯MetS-Z為極具預測力的風險指標。
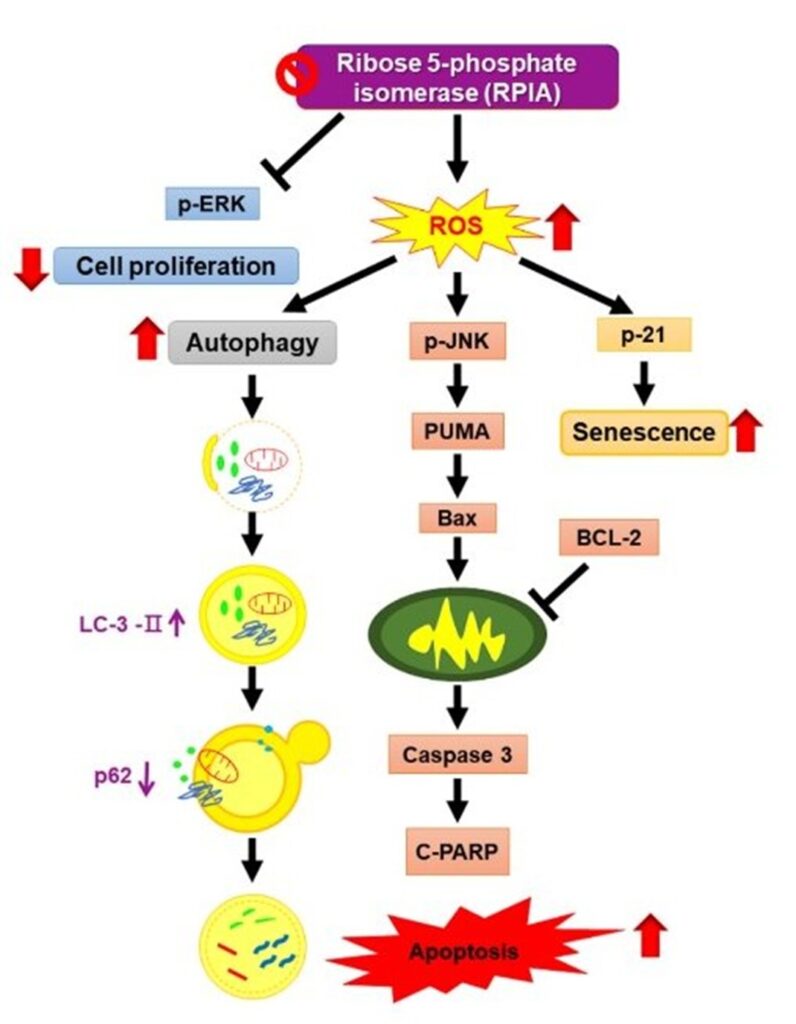
抑制5磷酸核糖異構酶A 誘導ROS活化肺癌中的自噬、細胞凋亡和細胞衰老
五磷酸核糖異構酶A(ribose-5-phosphate isomerase A, RPIA)是非氧化五碳糖磷酸途徑(pentose phosphate pathway)中的關鍵酶,不正常的五碳糖磷酸途徑過度活化與癌症生成有關。本院分子與基因醫學研究所喻秋華研究員與國立清華大學生命科學院生物科技研究所汪宏達教授團隊長期合作,探討RPIA在肝癌和大腸直腸癌中扮演的角色發現,RPIA在人類的肝癌、大腸直腸癌及肺癌的檢體中,相對於正常組織都有過量的表現量,並已共同發表RPIA調節肝癌和大腸直腸癌的腫瘤發生的機轉。然而,RPIA在肺癌中的作用仍不清楚;因此,研究團隊進一步探討抑制RPIA在肺癌中的作用及其潛在機制。
首先,研究團隊檢測人類肺癌中的RPIA蛋白發現,與正常組織相比,RPIA蛋白在人類肺癌有所增加;接著,在肺癌細胞中減弱(knockdown)RPIA基因表達則顯示自噬泡,且會增強吖啶橙(acridine orange)染色導致自噬體增加和產生GFP-LC3斑點,並顯示LC3-II量升高和p62量降低,共同表明在肺癌細胞中抑制RPIA活化了自噬作用。此外,RPIA的表達減少會藉由增加Bax的量、PARP的裂解及caspase3量來誘發更多的細胞凋亡,而且減少RPIA的表達會引發細胞衰老並增加肺癌細胞中p53和p21的量;重要的是亦提高了活性氧自由基(ROS)的量。最後,透過ROS清除劑N-乙酰-L-半胱氨酸(NAC)的處理可恢復在肺癌細胞中因RPIA表達減少所誘導的自噬、細胞凋亡和細胞衰老。總之研究顯示,減少RPIA表達會使ROS上升並活化肺癌細胞中的自噬、凋亡和細胞衰老。據研究團隊所知,這是關於在肺癌治療中的RPIA抑制的第一份報告。相關成果發表於Int J Mol Sci. 2022 Jul 17;23 (14):7883。
低濃度 4-ABP 促進人肝細胞和斑馬魚模型的肝癌發生
4-胺基聯苯(4-ABP)廣泛存在於偶氮染料、香菸煙霧中,是一種人類膀胱癌致癌物質。為了探討低濃度 4-ABP 是否誘導或促進肝癌發生並研究其潛在機制,國立中央大學生命科學系陳師慶特聘教授與本院分子與基因醫學研究所喻秋華研究員團隊合作,分別以人類肝臟細胞和斑馬魚肝癌模型探討低濃度4-ABP長期暴露誘發肝癌的可能機轉。
體外實驗證明,低濃度4-ABP重複暴露會通過 Ras/MEK/ERK 訊息傳遞途徑增加ROS,促進人類肝臟細細胞細胞增殖和遷移。動物實驗結果顯示,HBx、Src (p53-/-) 轉基因斑馬魚肝癌模型結果顯示長期暴露在低濃度4-ABP(1、10 和 100 nM) 會促使斑馬魚的肝癌進一步惡化,低濃度4-ABP誘導野生型斑馬魚在七個月形成肝癌 。證實4-ABP暴露為誘發肝癌的重要因子。
此外,我們在體外和體內觀察到 Ras-ERK 通路與 4-ABP 誘導的肝癌之間的相關性。我們的發現表明,低濃度的 4-ABP 重複暴露是肝癌的潛在危險因素。據我們所知,這是關於在 4-ABP 暴露後促進人類肝臟細胞增殖和遷移,並誘導斑馬魚肝癌形成的第一份報告。相關成果發表於Journal of Hazardous Materials(August 2021, 126954)。
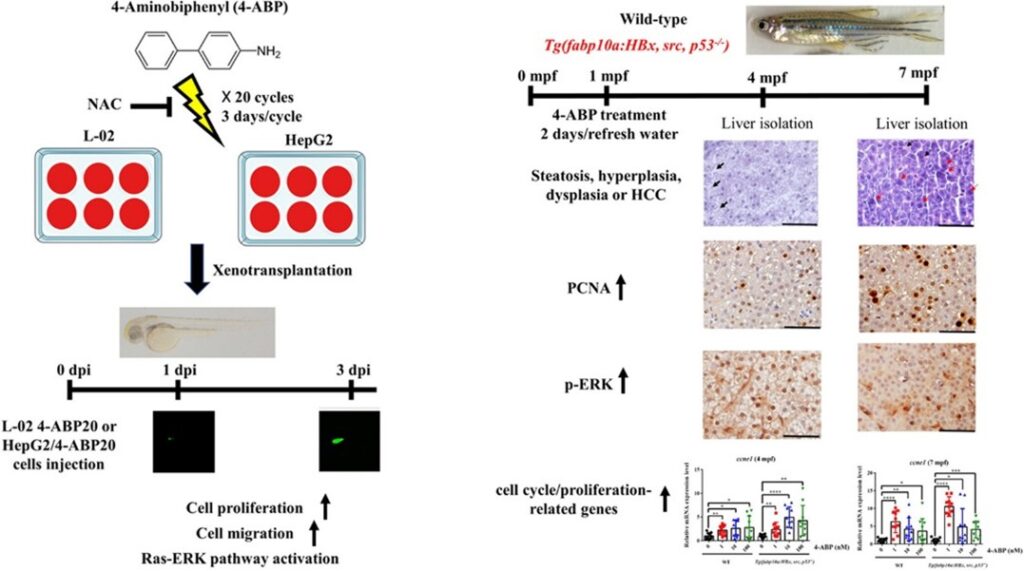 圖1. 低劑量的 4-ABP 重複暴露激活肝細胞中的 Ras-ERK 通路。 4-ABP 在肝細胞模型和斑馬魚模型中誘導或促進肝癌發生。
圖1. 低劑量的 4-ABP 重複暴露激活肝細胞中的 Ras-ERK 通路。 4-ABP 在肝細胞模型和斑馬魚模型中誘導或促進肝癌發生。
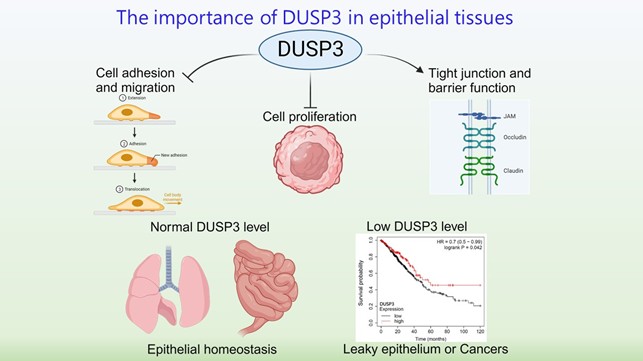
雙特異性去磷酸酶與上皮細胞癌
雙特異性去磷酸酶(DUSPs)最初被命名為MAPK去磷酸酶(MKPs),因為它們被發現是MAPKs的抑制分子。然而,近來研究發現,幾種DUSPs對於調控酪氨酸激酶信號傳導和多種細胞功能至關重要。分子與基因醫學研究所陳怡榮的團隊發現,一種非典型DUSP―DUSP3/VHR,在肺癌組織中的表達量會下降。DUSP3/VHR能夠去磷酸化ErbB受體(包括EGFR和ErbB2),並抑制肺癌細胞的腫瘤形成(J Biol Chem 2011)。DUSP3/VHR還抑制黏著斑激酶(FAK),而缺乏DUSP3/VHR的細胞在細胞黏附和遷移方面存在異常(Oncogene 2017)。最近,我們發現DUSP3/VHR調控緊密連接蛋白occludin(OCLN)的磷酸化引導的降解,並且DUSP3/VHR表現對於維持上皮細胞緊密連接和屏障功能是必需的(J Biomed Sci 2022)。我們的綜合研究成果顯示,DUSP3/VHR是調控上皮組織中增殖、黏附、遷移和細胞間連結的重要調節因子;DUSP3/VHR缺失在單層上皮細胞組織的癌症形成有重要角色。
IL-6R免疫治療結合標靶MCT-1/IL-6/CXCL7/PD-L1路徑,可預防三陰性乳癌的復發和轉移
三陰性乳癌(triple-negative breast cancer, TNBC)是乳腺癌治療的大挑戰,因其不具有雌激素受體(estrogen receptor, ER)、黃體素受體(progesterone receptor, PR)和人類表皮生長因子受體2(human epidermal growth factor receptor 2, HER2)而被歸類為“三陰性”,表現出強大的免疫抑制腫瘤微環境(tumor microenvironment, TME)以及高侵襲性和死亡率。相較於其他亞型的乳腺癌,TNBC目前的治療藥物的選擇較少,而免疫治療是一種新興的癌症治療模式,提供了TNBC治療的可能性。本院分子與基因醫學研究所徐欣伶研究員團隊長期深入研究TNBC的致病機制與治療策略,近年聚焦在免疫治療的潛力,特別在免疫逃逸、MCT-1訊息路徑、IL-6/IL-6R和CXCL7等因子的作用,以及序列性免疫治療策略(sequential therapy)的應用,發現結合IL-6R免疫療法及全身性標靶MCT-1/IL-6/IL-6R/CXCL7/PD-L1互連可增強免疫監視,進而抑制TNBC的侵襲性,是提高患者存活率的可行策略。此外,此項研究另一個重要發現為,過去標靶IL-6R主要用來治療類風濕性關節炎,她的研究發現提出可用作癌症之免疫治療。此篇論文是徐研究員於史丹佛大學進修期間與Michael P. Snyder教授合作,發表在Theranostics 2024;14(5):2167-2189。
MCT-1(multiple copies in T-cell malignancy, 也稱為MCTS1)是一個重要的腫瘤調節因子,在TNBC的發展中扮演著關鍵角色。過度表達的MCT-1可以促進EMT和TNBC的進展,並增強乳腺癌幹細胞(BCSCs)和M2巨噬細胞(促進腫瘤)的可塑性,因此MCT-1通路成為了潛在的治療靶點。已知MCT-1的多重拷貝是侵襲性乳癌的預後生物標記,MCT-1過度表現會刺激IL-6/IL-6R/gp130/STAT3軸,進而促進上皮間質轉化和癌症乾性。由於癌症幹性在很大程度上導致腫瘤轉移和復發,因此探討MCT-1和IL-6R的阻斷是否可以產生這些影響,並了解控制該過程的潛在機制。
徐研究員的研究發現,shMCT-1(MCT-1靜默)抑制TNBC細胞4T1中的發炎反應和轉移訊號的轉錄組,並抑制異種移植小鼠的腫瘤復發、轉移和死亡率。將IL-6R免疫療法和shMCT-1結合,則進一步減少腫瘤內M2巨噬細胞(會促進腫瘤)和 T調節細胞(Treg),並避免術後TNBC擴增; 此外,shMCT-1還可以增強IL-6R的免疫療法,有效預防術後TNBC轉移、復發和死亡。抗IL-6R可改善淋巴系統中的輔助性T細胞、細胞毒性T細胞和自然殺手(NK)細胞,並減少復發性和轉移性腫瘤中的Treg細胞。IL-6R 和 PD-L1聯合免疫療法比單一療法更大幅度的降低TNBC細胞幹性(stemness)和M2巨噬細胞活性。與同步治療相比,PD-L1和IL-6R的序列性免疫療法顯示出最佳的生存效果以及最低的術後復發和轉移,特別是在shMCT-1背景下。在TNBC細胞中, 她的團隊發現了MCT-1/IL-6/IL-6R/CXCL7/PD-L1軸的多個正向回饋迴圈(feedback loop),這增強了轉移微環境和免疫抑制微環境,假說示意圖請見圖一。臨床上,MCT-1/PD-L1/CXCL7 和CXCL7/IL-6/IL-6R高表現模式顯示乳癌患者預後及存活率較差。
TNBC的免疫治療前景充滿挑戰但也充滿希望。透過深入研究免疫逃逸機制、MCT-1訊息路徑及IL-6/IL-6R和CXCL7等因子的作用機制,應用序列性免疫治療策略,有望開創出更有效的TNBC治療模式。然而,這一領域仍需要進一步的研究和臨床驗證,以確保治療的安全性和有效性。徐研究員的研究結果為 TNBC 建立了一種創新的系統療法,可以提高臨床影響和患者存活率。
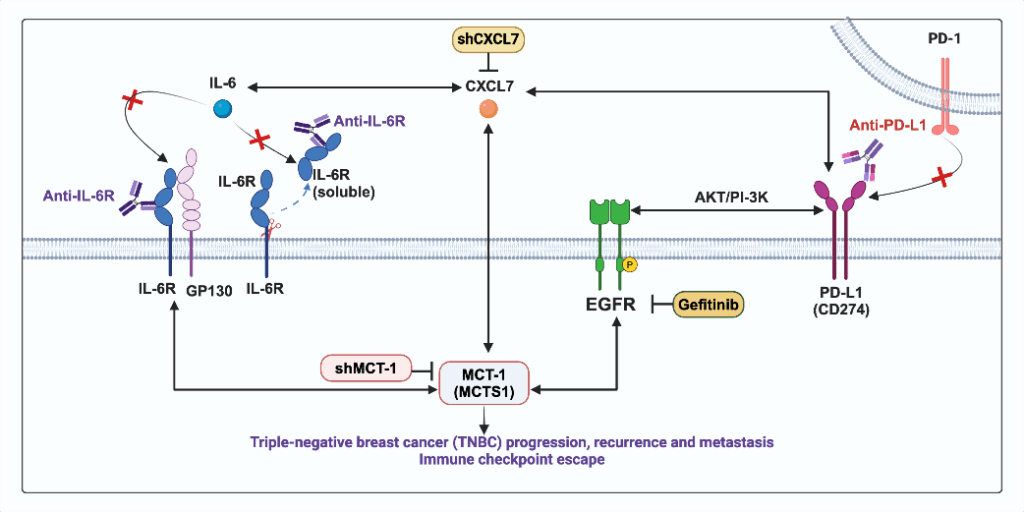
圖一.回饋迴圈假設示意圖 示意三陰性乳癌細胞中致癌蛋白MCT-1與 IL-6、CXCL7和PD-L1相互調控之關係。此圖以BioRender繪製IL-6/IL-6R/gp130 的複合形狀修改自BioRender 的Tocilizumab Humanized Antibody Against IL-6R模板)。
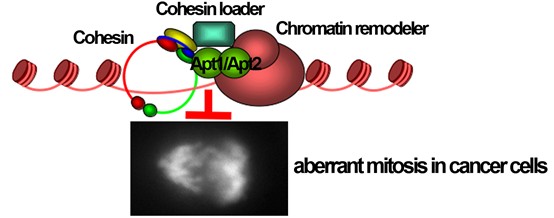
棕櫚酰水解酶非酵素活性之染色體黏連蛋白裝載功能
相較於正常細胞,癌細胞的基因體特別複雜,這些不正常的結構導致癌細胞在生長複製有絲分裂姐妹染色體分離時特別容易出錯。王紹文博士的實驗室以裂殖酵母為生物模式來研究這一現象。研究結果顯示前列腺癌細胞會藉由增加調控有絲分裂的染色體黏連蛋白(cohesin)與載體(loader)的表達來減緩有絲分裂所造成的壓力(mitotic stress)減少細胞的死亡。黏連蛋白需透過染色體重組複合體(RSC chromatin remodeling complex)的作用才能加載到染色體上。本研究證實在裂殖酵母中,該功能需要Phi1類棕櫚酰水解蛋白來橋接黏連蛋白與RSC複合體之間的交互作用。Phi1與黏連蛋白的Rad21和RSC複合體ATP水解酶Snf21相互作用,以促進黏連蛋白的染色體加載。在缺乏Phi1的狀況下,有絲分裂過程中,姊妹染色體提前分離,導致染色體分離滯後,與紡錘體監控點的激活。我們進一步證實人類Phi1同源蛋白Apt1與Apt2同樣具有染色體裝載的功能,而Apt1和Apt2與Rad21和人類Snf21同源蛋白Brg1的相互作用並不需要透過其酵素活性。與臨床證據一致的是Cohesin-Apt1-Brg1複合體在前列腺癌C4-2B細胞有過度表達現象,同時剔除Apt1與Apt2會導致這些細胞有絲分裂異常而死亡。這些數據顯示Cohesin-Apt1-Brg1複合體有抵抗癌細胞有絲分裂壓力促進細胞生存之功能。藉由臨床檢體的佐證,我們證實Apt1在細胞核內的高表達與前列腺癌患者的存活率呈現負相關,顯示Apt1可作為癌症檢驗的標記。前述結果發表於Nucleic Acid Research期刊2025第53期第gkaf257 頁。
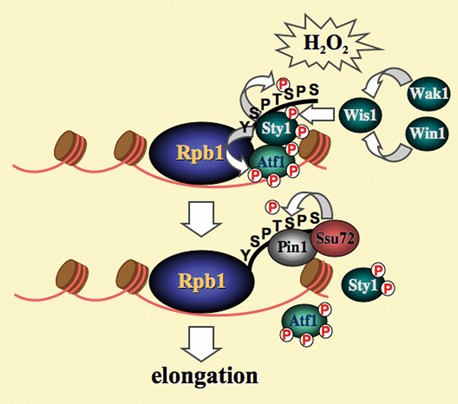
Pin1脯胺醯基異構酶調控癌細胞對氧化壓力的適應能力之分子機制
癌細胞由於代謝作用途徑的改變,不斷暴露在其所引起的氧化壓力下,如何有效反應調適降低氧化壓力所造成的毒性,對於癌細胞的存活相當重要。王紹文博士的實驗室以裂殖酵母為生物模式來研究細胞對氧化壓力的反應調適應變能力,意外發現Pin1脯胺醯基異構酶扮演了重要角色。Pin1普遍存在於真核細胞中,其主要的功能在催化脯胺酸胺基端肽鍵順/反構型的改變。比較特別而不同於其它脯胺醯基異構酶的是Pin1專一性的催化磷酸化酥胺酸或絲胺酸後所接的脯胺酸胺基端的順/反構型改變,可通過與RNA 聚合酶II (Pol II) 的最大組成分子Rpb1 C端結構區 (CTD) 的交互作用來調節 Pol II的結構和轉錄功能。我們證明了此功能在裂殖酵母細胞對氧化壓力的反應調適應變能力的重要性,並建立其調控的分子機制(如附圖)。在氧化壓力下,有絲分裂活化激酶 Sty1會被磷酸活化以促進氧化壓力反應解毒基因的表達。Sty1會磷酸化並穩定轉錄因子Atf1。Atf1將Sty1靶向特定的氧化壓力反應基因啟動子。錨定的Sty1通過與Rpb1-CTD 的直接鍵結徵募Pol II,並磷酸化位於七肽序列5處的絲氨酸。Pin1透過與絲氨酸5磷酸化CTD 的結合促成Sty1的解離,並直接募集Ssu72磷酸酶去磷酸化CTD以促進轉錄的延長作用增加解毒基因的表達。我們進一步證明人類Pin1亦具有相同的功能。Pin1在癌細胞中有過度表達的現象。Pin1的過度表達會增進細胞對氧化壓力的調適應變能力,促進癌細胞的成長與存活,以及影響化學療法中藉由氧化壓力產生細胞毒性之藥物療效,前述結果發表於Nucleic Acid Research期刊2021第49(2)期第805‐817 頁。
可用以進行藥物開發之皮膚脫色或發炎疾病的斑馬魚模式
色素細胞產生黑色素(melanin)之活性與膚色有關,同時也與皮膚紫外線的抵抗力與皮膚癌風險有關。黑色素,是由皮膚基底層的色素細胞中的黑素體(melanosome),經由酪胺酸酶(tyrosinase)之催化所產生。後天造成的色素脫失症(hypopigmentation)如白斑(vitiligo),是一種黑色素細胞(melanocyte)死亡的疾病,常伴隨有例如:巨噬細胞等免疫細胞的浸潤,在皮膚外觀上會有不規則白色的斑點、斑塊、灰髮,甚至有視網膜色素上皮層色素(retinal pigment epithelium, RPE)脫色、退化與視力受損等症狀發生。白斑的全球盛行率約為0.5 – 2%,其致病機制尚不清楚,因此無法提供有效的治療與預防,白斑並不危及性命,但由於影響外觀,可能對患者的心理造成負面影響。本院分子與基因醫學研究所江運金副研究員及其團隊成員許家豪博士後研究員,與中央研究院劉淦光博士合作,實驗證明nicastrinhi1384突變斑馬魚是一個極具潛力的脫色疾病模式,可應用於皮膚相關疾病的治療、開發脫色疾病或發炎藥物之研發。此論文已發表於國際知名皮膚研究期刊Journal of Investigative Dermatology(2020; 140:404-414)。
有關白斑的致病機制,已有一些相關研究發表:在基因組關聯性分析(GWAS)研究結果顯示,TYR以及幾個與自體免疫疾病相關的基因,例如:IL2RA和MHC均與白斑有關,在皮膚脫色發炎的位置,也可觀察到巨噬細胞。γ-secretase是由Presenilin、Pen2、Nicastrin所組成的複合蛋白,過去以γ-secretase 抑制劑進行白斑之人體臨床試驗,以及在實驗動物上,確實可改變毛色。有研究發現,缺失nicastrin會降低γ-secretase的穩定性,為深入探討其相關性,江運金副研究員利用nicastrinhi1384突變斑馬魚模式進行白斑的致病機制研究。研究團隊觀察到在nicastrinhi1384突變斑馬魚品系中,其子代在受精後(hours postfertilization, hpf)48小時,四分之一有色素脫失症,76 hpf則有八分之一呈現捲尾性狀(curled-up tail),這些突變所表現的漸進性色素脫失症可以很容易的在眼睛與表皮區域觀察到,並證明黑色素在nicastrinhi1384突變株的脫色現象是黑色素細胞死亡,並非基因表現之問題。
研究團隊以穿透式電子顯微鏡觀察nicastrinhi1384突變斑馬魚,量化因視網膜色素上皮層色素脫色所造成的黑素體變化情形。結果顯示,相較於野生種斑馬魚,nicastrinhi1384突變種的黑素體在受精3天後(3 days postfertilization, 3dpf)呈現多空泡的胞內體(multivesicular endosomes),其色素量亦少於野生種,顯示黑素體為未成熟狀態。nicastrinhi1384同型核子的粒線體(mitochondria),則呈現腫脹,其黑色素細胞呈現非細胞凋亡型的細胞壞死。過去的文獻曾提出,tyrosinase下游的代謝產物,例如:純黑色素(eumelanin),與黑色素細胞之壞死有關,研究團隊以tyrosinase抑制物-苯硫脲(phenylthiourea, PTU)抑制nicastrinhi1384突變斑馬魚tyrosinase之活性,在22 hpf一開始即施以PTU,所有子代在3 dpf皆可清楚觀察到表現dct基因黑色素細胞(dct-expression cell),而未施以PTU的nicastrinhi1384突變斑馬魚,則有四分之一的子代微量或幾乎不表現黑色素細胞標誌dct基因。接著在48 hpf再以PTU處理,雖已發生脫色,但顯示可以降低破壞黑色素細胞,並防止視網膜色素上皮層色素的脫色;同時,粒線體的腫脹以及黑色素細胞壞死的比例亦明顯降低。
此外,過去文獻提出nicastrin對γ-secretase作用在其基質之一的Notch並非必需,研究團隊以γ-secretase抑制劑N-[N-(3,5-difluorophenacetyl)-Lalanyl]-S-phenylglycine t-butyl ester (DAPT) 處理野生種斑馬魚4體節(somite stage)的胚胎,呈現不成熟的黑色素細胞與腫脹的粒線體,亦觀察到含有破碎黑素體的壞死黑色素體與粒線體皺褶減少;對野生種幼魚施以DAPT可觀察到脫色;對48 hpf的胚胎施以DAPT與PTU,可防止脫色(圖1)。以DAPT處理nicastrinhi1384突變斑馬魚,則會強化脫色。這些實驗結果顯示,nicastrinhi1384突變斑馬魚tyrosinase依賴性的脫色,是因為γ-secretase活性的降低。此外,過去的研究指出,白斑病的黑色素細胞壞死與粒線體壓力,會驅動斑馬魚的先天性免疫反應,並接續產生後天免疫的分子機制。研究團隊進一步測試nicastrinhi1384突變斑馬魚之先天免疫系統是否處於活化狀態,透過植入具有報導功能的巨噬細胞(macrophage reporter line, Tg(mpeg1:mCherry)gl23)發現,巨噬細胞會在黑色素細胞所在之處,並吞噬黑色素細胞,而且,已經受損的黑色素細胞會被鄰近的黑色素細胞所吞噬,顯示在nicastrinhi1384突變斑馬魚模式下,黑色素細胞損傷會活化先天性免疫反應。
綜合上述,研究團隊發表了nicastrin在白斑機制上的嶄新功能。當明顯降低nicastrin之基因轉錄,影響黑素體之成熟,造成tyrosinase依賴性粒線體腫脹與黑色素細胞死亡,nicastrinhi1384突變斑馬魚模式證實,皮膚脫色的表現是因為γ-secretase失去活性的結果。此外,穿透式顯微鏡影像結果顯示,巨噬細胞會召集以吞噬黑色素細胞碎片,詳見圖2示意圖。此研究提出nicastrinhi1384突變是一個有潛力的斑馬魚脫色疾病模式,可用於皮膚相關疾病的治療、開發脫色疾病或發炎之藥物。
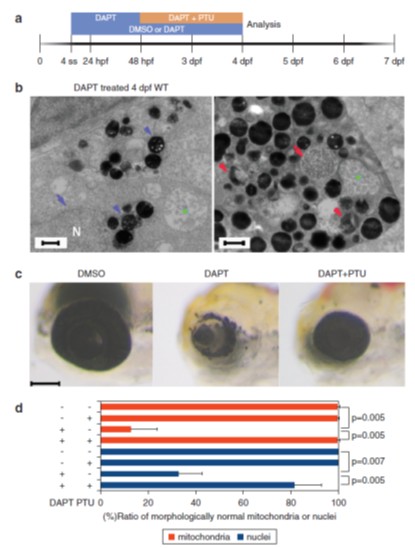
圖1(a)以DAPT或DAPT+PTU處理的時間;(b)當以DAPT處理,野生種幼魚呈現黑色素細胞不成熟(藍色箭頭),粒線體皺褶減少(綠色箭頭);(c)以DAPT處理野生種斑馬魚,有脫色現象;但同時以DAPT+PTU處理,則還原脫色;(d)粒線體與細胞核的定量統計
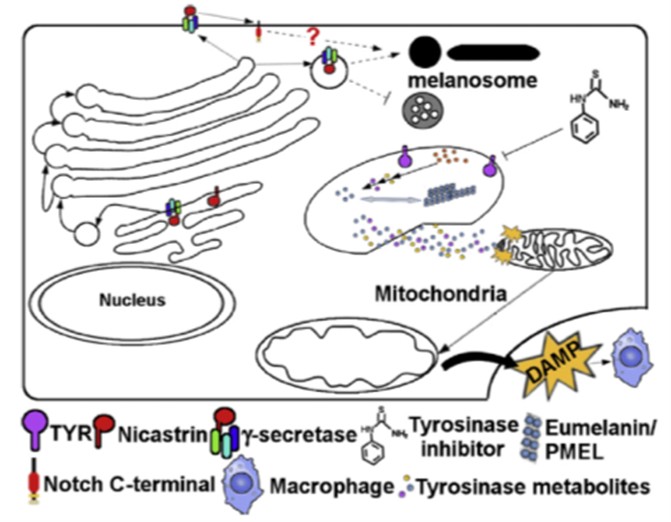
圖2:nicastrin在白斑機制之示意圖。Nicastrin為γ-secretase的成員之一,維持黑素體所必需的蛋白,nicastrin減少會損害γ-secretase的功能,接續干擾黑素體的發育。之後,黑素體缺陷導致釋放出黑素體毒性物質,例如:eumelanin及其前驅物會傷害胞器,例如:粒線體。此嚴重的傷害觸發黑色素細胞的壞死和導致DAMP(damage-associated molecular pattern)的釋放,隨後會召集例如:巨噬細胞(吞噬細胞),吞噬受損的黑素細胞並呈現抗原
斑馬魚用於研究家族性滲出性玻璃體視網膜病變
家族性滲出性玻璃體視網膜病變(Familial exudative vitreoretinopathy, FEVR)是一異源性(heterogeneous)的遺傳疾病,其特徵是異常血管形成於周圍視網膜,導致視網膜脫離和嚴重的視力障礙。臨床上發現病人有家族內的病徵差異及雙眼間的病徵嚴重性不對稱等特徵。至今已有五個基因被鑑定及驗證,它們的缺失會造成FEVR:FZD4,LRP5,TSPAN12,NDP和ZNF408。前四個已被證明參與Norrin (NDP基因的產物)誘導的FZD4/β-catenin訊息通路。ZNF408的參與機制則還不是很清楚。雖然如此,只有大約50%的病人,他們的病變是由於這五個基因中的一個的缺失所造成。另外的50%的病人其病因則未知。
陽明大學生命科學系暨基因體科學研究所的鍾明怡老師及台北榮總的陳世真主任經由分析FEVR病人檢體、家族史、外顯子測序(exome sequencing)發現了在RCBTB1 (Regulator Of Chromosome Condensation And BTB Domain-Containing Protein 1, 人源全長重組蛋白)基因上的變異可能是造成FEVR的原因。進一步用細胞培養及分生的方法,證明了RCBTB1對Norrin或Wnt3a (Wingless-Type MMTV Integration Site Family, Member 3A, 其轉錄之蛋白與特發性青少年骨質疏鬆症及顱骨發育不良有關)誘導的Wnt/β-catenin訊息通路激活是必需的。江運金老師研究團隊(助理朱國彰實際操作相關實驗)與陽明大學及台北榮總團隊合作使用斑馬魚驗證了rcbtb1的表現減量(使用反股寡核苷酸morpholino,抑制蛋白轉譯)會造成視網膜的血管病變(圖一)。進一步我們更證明了rcbtb1與ndp兩基因的缺失對視網膜的血管病變有加成作用(圖二),表示遺傳上rcbtb1能與Norrin誘導的Wnt/β-catenin訊息通路一起作用。這些結果與由細胞培養研究中所獲得的結果是相符合的,並且可以互相印證。為了證明基因減量(knockdown)的專一性,我們同時注射人類RCBTB1的mRNA與rcbtb1的morpholino於斑馬魚胚胎中發現表型可回復得比較正常;相反地,病人的變異RCBTB1 mRNA就無拯救的效果。我們也使用了另一種方法(CRISPR interference,抑制基因轉錄)來達成基因減量的功能,胚胎同樣有視網膜的血管病變。這些實驗顯示了我們用的基因減量法是專一的而且沒有脫靶(off-target)效應。這篇論文以共同通訊作者發表於2016的Human Molecular Genetics。
我們已自英國進口了rcbtb1缺失的變異種(英國的Sanger Institute有一巨大的變異種收集庫,可查Zebrafish Mutation Project;也可由TALEN或CRISPR/Cas9的技術來產生變異種)。待它們長成成魚,我們將可進一步研究,如在成魚視網膜的血管表型;用基因微陣列(microarray)去發現轉錄體(transcriptome)的差異,可提供Rcbtb1如何參與Norrin誘導的Wnt/β-catenin訊息通路的線索;也可用此變異種去篩選能讓視網膜血管長好的小分子藥物等。
總之,由於斑馬魚具有胚胎透明、體外受精、體外發育、繼代快、成本低、有許多有利研究的轉殖魚和變異種可用及多種基因操作技術成熟等優點且基因組與人類有87%的高度同源性,是研究人類遺傳疾病的另一種可選擇且方便使用的動物模式。
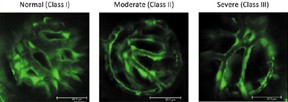 圖一. 以morpholino達成斑馬魚胚胎基因減量,探討rcbtb1的表現對視網膜的血管病變之影響
圖一. 以morpholino達成斑馬魚胚胎基因減量,探討rcbtb1的表現對視網膜的血管病變之影響
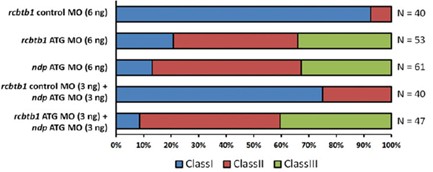 圖二. 以半定量分析,探討不同基因組合之缺失對視網膜的血管病變之影響
圖二. 以半定量分析,探討不同基因組合之缺失對視網膜的血管病變之影響
未經歷新冠病毒之川崎氏症兒童血清對新冠病毒仍具有體免疫交互保護
川崎氏症(Kawasaki disease, KD)是最常見的兒科系統性血管炎疾病,目前的標準治療是靜脈注射免疫球蛋白,有5%至20%的患者在急性期發生冠狀動脈病變(coronary arterial lesions, CAL)。2019年全球爆發新冠病毒(SARS-CoV-2)大流行,可導致嚴重特殊傳染性肺炎(COVID-19),但全球皆發現某些兒童在感染SARS-CoV-2後,會發生身體多系統的發炎性疾病,通稱「兒童多系統炎症綜合症(Multisystem Inflammatory Syndrome in Children, MIS-C)」,其臨床症狀與KD非常相似,來自義大利的首發報告中,發現類川崎氏症疾病的發病率異常增加了約30倍之多。基於KD與MIS-C的臨床表現高度相似,本院分子與基因醫學研究所劉鴻興醫師與台大醫院合作,評估是否KD患者對冠狀病毒之體免疫或有”漏洞”,方導致MIS-C及KD兩種類似疾病的產生。由於台灣嚴密的防疫措施,防止了SARS-CoV-2 Omicron變異株發生前的大爆發,利用這樣的環境條件,可評估未曾經歷新冠病毒之川崎氏症兒童血清,對新冠病毒之體免疫保護效果是否有所差異。此研究以2019年之前(29例)及2019-2020年期間未曾接觸過SARS-CoV-2的KD兒童血清(74例),及發燒(24例)或未發燒(54例)的其他兒童血清當作控制組,測試對SARS-CoV-2原始株的抗體中和反應。初步結果顯示,這些不同年份血清樣本對SARS(嚴重急性呼吸道症候群冠狀病毒)、SARS-CoV-2 (即nCoV, novel coronavirus)、MERS(Middle East respiratory syndrome,中東呼吸症候群冠狀病毒)等三種病毒的spike protein交叉反應類似(圖1A),大多數樣品對所有三種病毒spike protein要不均呈陽性結果,或者都沒有交叉反應。研究中更進一步評估對SARS-CoV-2有反應之抗體,是否有足夠之病毒中和能力,發現不論是控制組或KD族群,相較WHO標準血清檢體,大多皆有至少符合WHO的低(low)中和能力(圖1B)。這些交叉反應的結果,很可能是由其他常見冠狀病毒引起的,例如已經存在社區中的人類冠狀病毒229E、OC43或NL63,且一定程度上能遏制SARS-CoV-2原始株對KD患者之感染,這與我們最初的假設相矛盾。儘管我們在KD患者血清中沒有發現冠狀病毒特異性的“漏洞”,但仍然有可能體液免疫對於尚未識別的其他抗原反應性不同,或者在某些情況下,冠狀病毒特異性體免疫可能會被不適當地增強。總之,本研究證實在台灣未曾接觸SARS-CoV-2的KD兒童體免疫,同樣能提供SARS-CoV-2的交叉保護作用。相關研究成果已發表於2023年的《臨床微生物學與感染學雜誌》(Clinical Microbiology and Infection 29 (2023) 257.e1-257.e5)
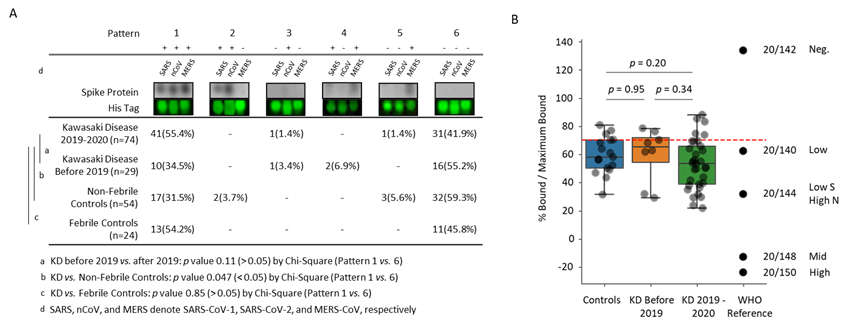
圖1、不同年份血清樣本 (A)與SARS、nCoV、MERS進行micro western分析結果 (B) 及與WHO標準檢體之中和能力比較。
從海洋天然物中發現並開發具口服活性的選擇性STING抑制劑
Excavatolide B (excB) 是從台灣的軟珊瑚中提取的海洋蘆薈烯類二萜化合物。其抗發炎特性和獨特的電親性環氧內酯官能基結構,促成了由宋秉鈞博士、鄒倫博士及張明姿博士所領導的跨機構、跨學科合作,致力於揭示excB的細胞靶點與作用機制。團隊利用來自軟珊瑚 Briareum stechei培養物的大量excB來源,並開發了功能性excB探針,透過化學蛋白質組學策略確定了STING (Stimulator of Interferon Genes) 是excB在哺乳動物細胞中的直接靶點。透過電腦分子對接模擬、與已知STING抑制劑競爭試驗、生化分析、基因學驗證和質譜技術,研究證明excB可選擇性地與STING膜附近的半胱氨酸 (Cys91) 殘基共價結合,並透過其環氧內酯官能基阻止STING的S-棕櫚酰化,進而抑制STING信號傳導及其介導的第一型干擾素反應。這項研究成果已於2023年發表在《Communications Chemistry》,揭示了一類新型的共價STING抑制劑,為未來針對這個在癌症、自體免疫和慢性炎症疾病中驅動發炎反應的重要先天免疫調節因子的抑制劑設計提供了新方向。
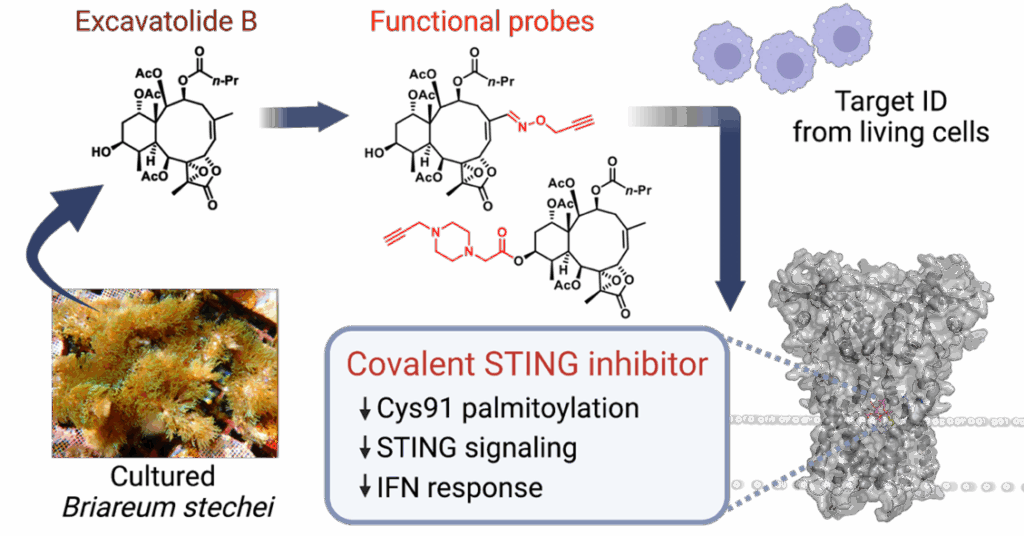
海洋二萜類化合物靶向哺乳動物細胞中的STING棕櫚酰化. 2023年發表於《Communications Chemistry》,第6卷,第153篇文章。此研究首次揭示了蘆薈烯類(二萜類化合物)的直接細胞靶點,並發現了一個新型共價STING抑制劑類別,為開發針對STING相關疾病的創新療法提供了重要線索。
新型STING抑制劑結構類別的發現,激發了同一團隊進一步優化excB骨架,以提升其STING抑制活性和藥理特性。在保留環氧內酯等關鍵功能結構的前提下,團隊合成並篩選了近百種excB衍生物,最終確定GHN105——一種具口服活性、選擇性標靶STING的共價抑制劑。透過excB的晚期結構多樣化修飾,GHN105引入了提升溶解度的功能基團,同時增強了對多種人類STING變體(包括導致遺傳性自身免疫疾病的致病性S154變體)的共價抑制活性。GHN105保持了對STING的Cys91殘基的高度選擇性,並在細胞和動物模型中呈現劑量依賴性地抑制STING信號傳導及干擾素反應。
口服給藥的GHN105在體內成功與STING結合,並在急性結腸炎小鼠模型的延遲治療中顯著逆轉了關鍵病理特徵。這項研究成果將於2025年發表在《Journal of Medicinal Chemistry》,報導了首款具口服活性的STING抑制劑,並在臨床前疾病模型中證實了其治療效果,突顯了大環蘆薈烯骨架在藥物開發中的巨大潛力。
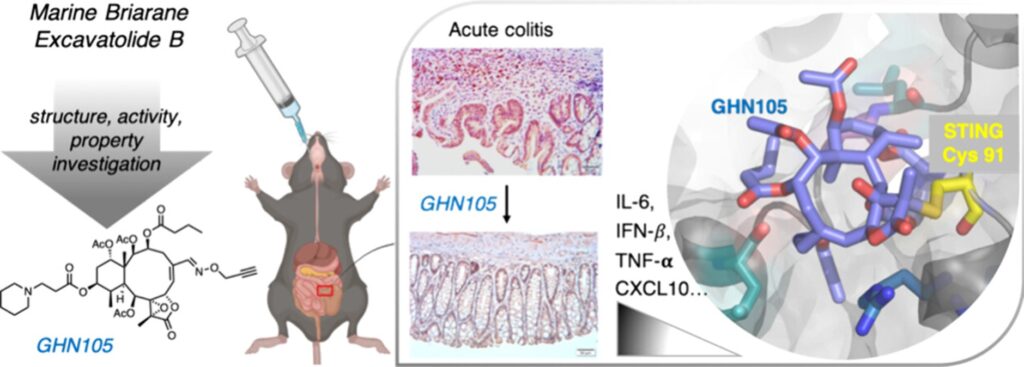
源自大環海洋二萜類化合物的口服可吸收、位點選擇性共價STING抑制劑。 2025年發表於《Journal of Medicinal Chemistry》,第68卷,5471-5487頁。本研究顯示,合成的蘆薈烯類衍生物GHN105是一種安全、具有位點選擇性且口服活性的共價STING抑制劑,在結腸炎的臨床前動物模型中展現了顯著的治療效果。
硝基脂肪酸的細胞靶點
硝基脂肪酸(nitro-fatty acids, NFAs)是一種來自飲食或內源性產生的親電子的脂質介質,而研究結果指出硝基脂肪酸在多種發炎反應和纖維化疾病模式中,具有良好的抗發炎及細胞保護效果;其中,以10-硝基油酸(10-nitro-oleate, CXA-10)作為抗發炎的治療藥物正在進行二期臨床試驗。硝基脂肪酸主要的功效來自於它們與蛋白質形成共價化合物的能力,目前僅少數蛋白已證實可被硝基烷基化(nitro-alkylated),但其細胞靶點的涵蓋範圍仍未被完整揭示。
本院分子與基因醫學研究所張明姿助研究員、生技與藥物研究所鄒倫副研究員與長庚大學生物醫學系陳怡婷副教授跨領域合作,結合生物正交化學與化學蛋白質體學,首次於人類活體細胞中全面性探索硝基脂肪酸的靶點(圖),成功在活體THP1巨噬細胞中鑑定出多達184個硝基烷基化蛋白質(nitro-alkylated proteins)。當中除了已知的蛋白質靶點如stimulator of interferon genes(STING)、kelch-like ECH-associated protein 1(KEAP1),多數為硝基脂肪酸的新靶點,其中更涵蓋了發炎相關調控因子及脂質代謝、運送等重要蛋白質如signal transducer and activator of transcription 3(STAT3)、toll-like receptor 2(TLR2)、retinoic X receptor alpha(RXRα)和glucocorticoid receptor(NC3R1)。此外,研究團隊進一步驗證硝基油酸酯通過特定位點的共價修飾調控NR3C1的功能。該研究大幅擴展了活體細胞中硝基脂肪酸細胞靶點的研究範疇,並為進一步瞭解硝基脂肪酸作為信號分子或多靶點治療劑的作用機制提供了重要的基礎。相關成果已發表於Redox Biology(2021 October, 102126)。
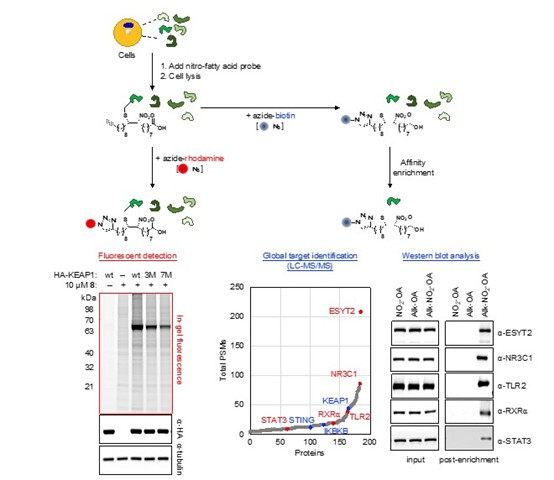
圖:結合生物正交化學與化學蛋白體學剖析複雜的人類蛋白質組,找出硝基脂肪酸的細胞靶點。運用帶有獨特官能基的硝基脂肪酸化學探針和隨後連接的可視或親和標誌,可以對硝基烷基化的蛋白質進行螢光檢測、純化和全面鑑定。
長鏈非編碼RNA LncHIFCAR可望開發為口腔癌生物偵測標記與治療標靶
腫瘤長久以來是醫師束手無策的棘手難題。目前已知”缺氧”是大多數腫瘤快速生長下的微環境特徵,更是引發癌細胞後續惡性化、抗藥性與轉移的重要原因。缺氧腫瘤不易治療且病患預後狀況較差,然而現今對於腫瘤缺氧反應之相關機轉並未完全明瞭。本院分子與基因醫學研究所龔行健榮譽研究員與北醫大施景文博士、柳營奇美醫院蔣維凡醫師共同組成之跨校跨領域研究團隊,結合基礎科學及臨床醫療研究,成功揭示一在口腔癌細胞大量表現之長鏈非編碼RNA (long non-coding RNA; lncRNA) LncHIFCAR (long noncoding HIF-1a co-activating RNA 長鏈非編碼HIF-1a 輔激活RNA),正是腫瘤缺氧反應的重要因子,更為促使口腔癌惡化轉移的致病關鍵。不同於以往癌症研究著重於探討細胞蛋白或微小RNA相關之致癌機轉,此一研究突破性地發現細胞中之長鏈非編碼RNA與腫瘤缺氧反應有密切關連,不但再次證實這類RNA在調控基因表達的功能角色,更由於該RNA之生化特性,LncHIFCAR可望成為未來口腔癌之診斷標記與治療標靶,有助於口腔癌病患的治療選擇及抑制腫瘤轉移標靶藥物的研發。
口腔癌是全世界人類第五大癌症。在台灣,由於嚼食檳榔的特殊文化,口腔癌的發生率居高不下,排名十大癌症死因的第四位,同時也是發生率和死亡率成長最快的癌症之一。口腔癌不但常發生於青壯年時期,且男性發生率是女生9倍之多。近年來,罹患口腔癌之年齡層有逐年下降的趨勢,不僅是國人健康重大課題,更突顯出發展口腔癌早期診斷之生物指標以及有效治療之醫療策略的急迫性。包含口腔癌在內的許多腫瘤,常因快速生長導致氧氣供應不足,內部呈現缺氧狀態。缺氧是決定腫瘤惡性化發展的關鍵因素,近年來發現,腫瘤細胞在缺氧環境下,會激發【缺氧誘導因子-1a】(HIF-1α; Hypoxia-inducible factor-1α)的活性,HIF-1α進一步與許多轉錄輔助因子結合形成完整的HIF-1複合體,坐落於一系列相對應之缺氧反應基因上,進而啟動此一系列缺氧反應相關基因群之表現,以幫助腫瘤細胞於缺氧環境下存活,甚而促進癌細胞轉移、惡化與抗藥性。因此,詳細了解腫瘤細胞調控缺氧反應的機轉是科學界與臨床醫學急切的課題。
長久以來,人們深信經由生物中心法則 (Central dogma;亦即DNA製造RNA,RNA製造蛋白質) 製造出來的蛋白質是執行生物功能的基本單元;然而,近年來由於高通量DNA定序技術的快速發展而發現許多”非編碼RNA (non-coding RNA)”,其本身不具有編碼和轉譯成蛋白之能力,因此又被暱稱為生物學中的暗物質。長鏈非編碼RNA(lncRNA)即為其中之一大類,雖然種類眾多且數量龐大,我們對其了解相當有限,但目前越來越多的研究證據顯示,這些暗物質在基因表現調控與許多致病機轉上扮演關鍵角色。也因此,研究團隊以長鏈非編碼RNA為研究主題,發現缺氧誘發的長鏈非編碼RNA LncHIFCAR,可做為HIF-1α的輔激活因子。此一RNA分子在細胞遇到缺氧狀況時大量表現,並和HIF-1α直接結合,幫助聚集其他轉錄輔助因子而活化缺氧反應基因群的表現,進而幫助腫瘤細胞在缺氧環境中調整代謝途徑、有效存活、快速增生,並促進腫瘤侵襲與轉移,造成癌症惡化 (圖一)。LncHIFCAR不但是目前首次發現具輔激活HIF-1α能力之長鏈非編碼RNA,更由於其可以利用病患血液進行偵測,因此具有高度臨床應用價值。
值得注意的是,研究團隊進一步對柳營奇美醫院所收集的國內口腔癌患者的組織樣本進行分析。結果發現,相對於周邊的正常組織,腫瘤組織內的LncHIFCAR 有大量表現的趨勢。進一步對LncHIFCAR的表現作分群,LncHIFCAR的表現量高的口腔癌患者其三年內的 腫瘤復發存活率 (disease-free survival rate)與 整體存活率 (overall survival rate)均顯著較LncHIFCAR的表現量低的口腔癌患者為差 (圖二)。生物統計分析更發現LncHIFCAR的病理組織表現量可作為口腔癌患者腫瘤復發率的獨立預後指標,顯示LncHIFCAR的表現量於臨床診斷上的應用潛力。研究團隊更以動物模型進行試驗,分別將一般的口腔癌細胞株與降低LncHIFCAR 表現的口腔癌細胞株經由尾部靜脈注射至小鼠體內。結果發現,降低LncHIFCAR 表現的口腔癌細胞株轉移至小鼠肺部的能力顯著降低,進一步證實LncHIFCAR 於腫瘤轉移的關鍵角色以及其作為癌症治療標靶的深厚潛力 (圖三)。
此一研究由國衛院與北醫大、柳營奇美醫院共同合作而展現突破性成果,研究成果深受重視,於2017刊登於國際知名期刊《自然通訊》 (Nature Communications) 上。近年來,以HIF-1α蛋白表現量作為預後根據與治療標靶的想法與策略遭受諸多困難與爭議,原因可能在於HIF-1α的整合性調節角色。基於RNA本身的生化特性,聯合研究團隊所發現可做為HIF-1α的輔激活因子的長鏈非編碼RNA LncHIFCAR,可望設計成更為靈敏簡便之生物標記檢測套組,以做為早期篩檢或預測口腔癌惡化及轉移的基礎。此外,更可進一步設計及開發針對LncHIFCAR RNA 專一性的阻斷性分子,透過微調LncHIFCAR RNA之表現量,作為針對具有高度惡化潛力口腔癌病患之精準治療方式。此一嶄新的分子指標不但能作為個人化癌症治療的有效策略,同時將提供未來口腔癌治療流程設計的依據,可望為國人的健康提供更多助益。
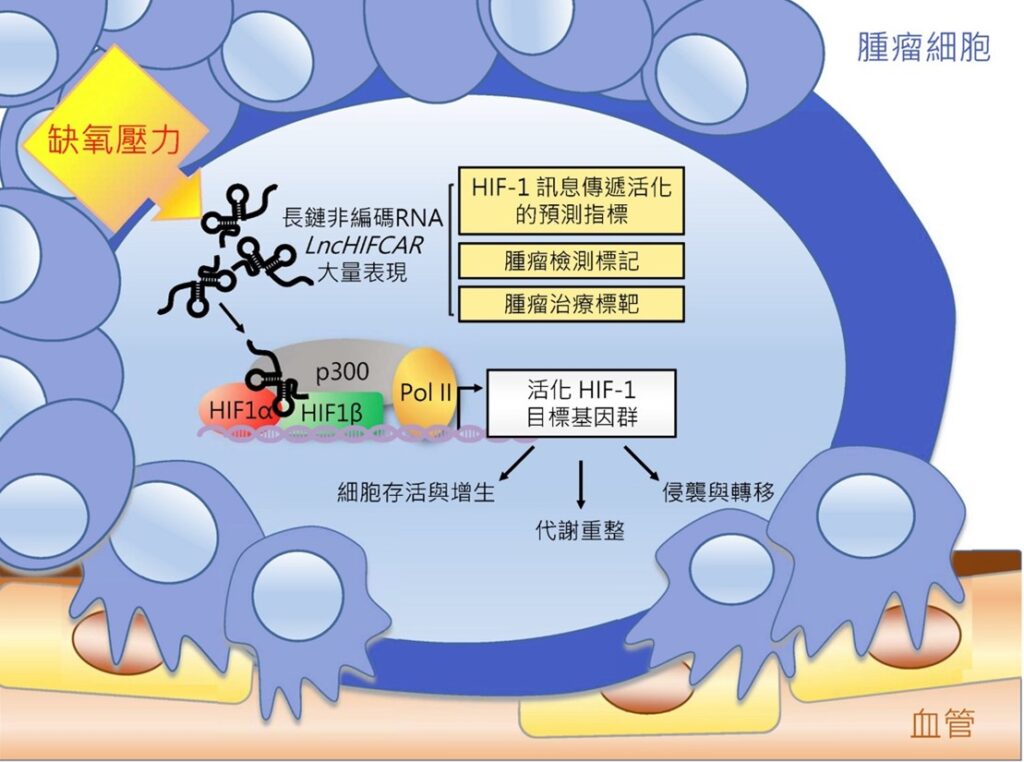 圖一: 長鏈非編碼RNA LncHIFCAR 與HIF-1 訊息傳導在腫瘤形成與轉移過程所扮演之角色模型。於快速生長的腫瘤細胞中,缺氧所誘發的長鏈非編碼RNA LncHIFCAR,為HIF-1α的輔激活因子,藉由和HIF-1α的直接結合,LncHIFCAR可幫助聚集其他轉錄輔助因子以形成HIF-1轉錄複合物而活化缺氧反應相對應的HIF-1目標基因群的表現,進而幫助腫瘤細胞在缺氧環境中進行代謝重整、有效存活、快速增生,並促進腫瘤侵襲與轉移,造成癌症惡化。
圖一: 長鏈非編碼RNA LncHIFCAR 與HIF-1 訊息傳導在腫瘤形成與轉移過程所扮演之角色模型。於快速生長的腫瘤細胞中,缺氧所誘發的長鏈非編碼RNA LncHIFCAR,為HIF-1α的輔激活因子,藉由和HIF-1α的直接結合,LncHIFCAR可幫助聚集其他轉錄輔助因子以形成HIF-1轉錄複合物而活化缺氧反應相對應的HIF-1目標基因群的表現,進而幫助腫瘤細胞在缺氧環境中進行代謝重整、有效存活、快速增生,並促進腫瘤侵襲與轉移,造成癌症惡化。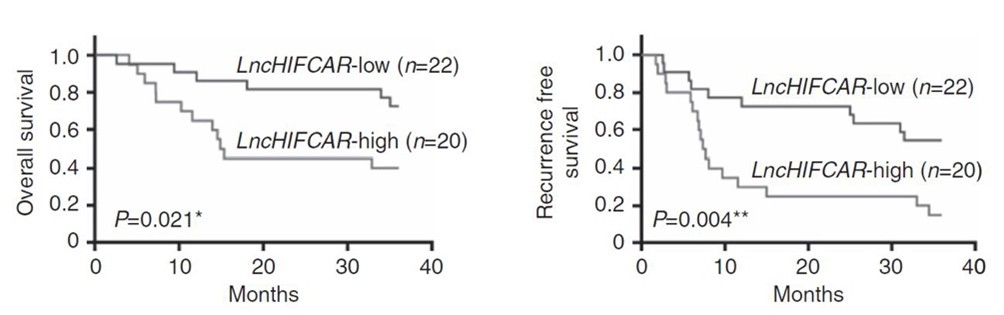 圖二: 長鏈非編碼RNA LncHIFCAR 表現量與42位口腔癌病人資料的追蹤分析。
圖二: 長鏈非編碼RNA LncHIFCAR 表現量與42位口腔癌病人資料的追蹤分析。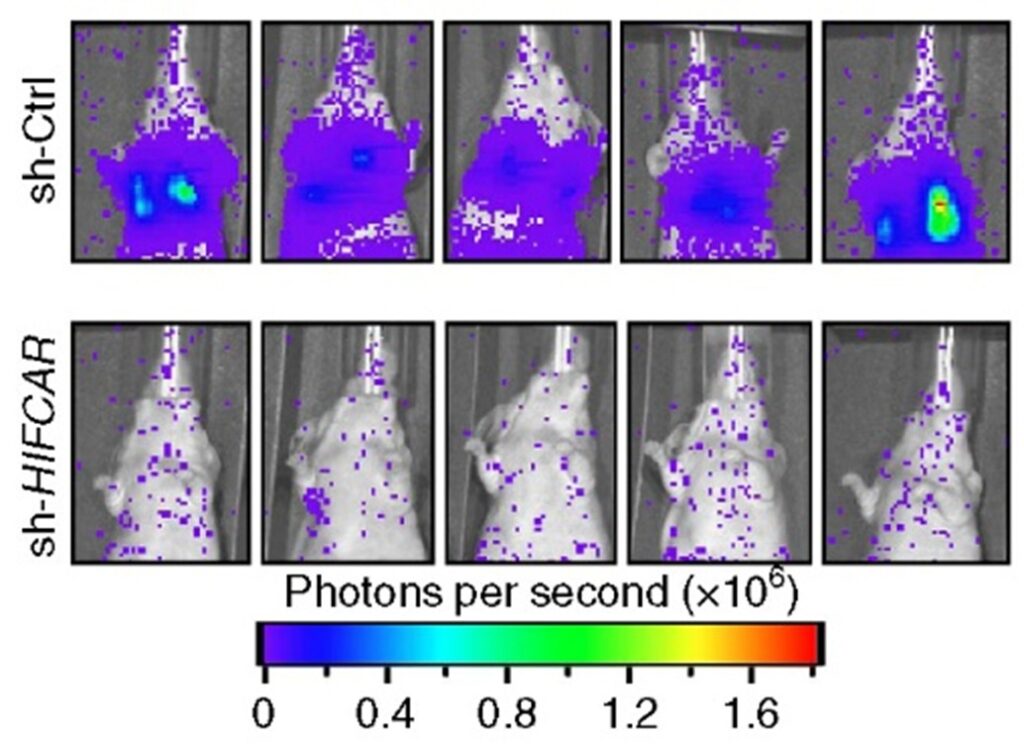 圖三: 降低LncHIFCAR 之表現能有效減少口腔癌細胞的肺轉移能力。分別將控制組的口腔癌細胞株(sh-Ctrl)與降低LncHIFCAR 表現的口腔癌細胞株(sh-HIFCAR)經由尾部靜脈注射至小鼠體內。結果發現,降低LncHIFCAR 表現的口腔癌細胞株轉移至小鼠肺部的能力顯著降低
圖三: 降低LncHIFCAR 之表現能有效減少口腔癌細胞的肺轉移能力。分別將控制組的口腔癌細胞株(sh-Ctrl)與降低LncHIFCAR 表現的口腔癌細胞株(sh-HIFCAR)經由尾部靜脈注射至小鼠體內。結果發現,降低LncHIFCAR 表現的口腔癌細胞株轉移至小鼠肺部的能力顯著降低
KDM4A激活E2F1調節PDK依賴性的粒線體氧化和糖解的代謝轉換
組蛋白賴氨酸甲基化 (histone lysine methylation) 經常參與轉錄調節,組蛋白賴氨酸脫甲基酶 (histone lysine demethylase, KDM) 是特異性催化從賴氨酸殘基去除甲基的酶。自自第一個KDM發現至今,已經知道有許多的KDM在不同癌症有遺傳上的改變或異常的表現,因而KDM被認為Targeting KDMs thus has been increasingly recognized as an anticancer therapeutic strategy ( Hojfeldt et al., 2013 ; McGrath and Trojer, 2015 ).是抗癌治療策略的研究方向。如同其他KDM4家庭成員,KDM4A被認為是基因激活或抑制的轉錄調節因子,在許多癌症類型,包括前列腺癌,乳腺癌,肺癌和結腸癌的過度表達,是細胞增殖的關鍵。龔行健特聘研究員研究團隊過去的研究提出龔行健特聘研究員的,在LNCaP前列腺癌細胞,以小分子抑製劑NSC636819選擇性抑制KDM4A/4B,有顯著的生長遲緩及增加細胞凋亡 (Chu et al., 2014)。
E2F1 is well recognized as a partner of retinoblastoma protein (Rb) and as a critical factor in growth regulation by serving as a transcriptional activator of many cell cycle genes.E2F1被認為是視網膜母細胞瘤蛋白(retinoblastoma protein, Rb)的參與者,並且是許多細胞週期基因的轉錄激活子生長調節的關鍵因素。There is strong evidence that E2F1 is involved in the development of CRPC.強有力的證據闡明E2F1與CRPC (去勢無效性前列腺癌, castration-resistant prostate cancer) 的發展有關,For instance, E2F1 overexpression leads to castration-resistance phenotype of LNCaP ( Libertini et al., 2006 ), E2F transcriptome is one of the prominent molecular signatures of CRPC( Sharma et al., 2013 ), and E2F1 coordinates with AR ( Ramos-Montoya et al., 2014 ) to modulate genes involved in CRPC.The mechanism however, remains obscure.然而作用機制尚不清楚。
Recently, tumor metabolism has gained increasing attention in cancer research.腫瘤代謝在近期的癌症研究獲得越來越多的關注。One of cancer cell’s hallmarks is the Warburg effect, by which cancer cells heavily rely on glycolysis to obtain energy and produce macromolecules that are required to sustain rapid proliferation.癌細胞的特點之一是Warburg效應(Warburg effect),癌細胞重度透過糖解(glycolysis)獲得能量,並製造用以維持快速增殖的大分子。To this end, cancer cells divert the glycolytic metabolite pyruvate from entering mitochondria for the tricarboxylic acid (TCA) cycle, and suppresses oxidative phosphorylation, allowing cancer cells to avoid excessive production of reactive oxygen species (ROS) and prevents mitochondria-driven apoptosis ( Cairns et al., 2011 ; Dang, 2012 ; Schulze and Harris, 2012 ).為此,癌細胞轉移進入線粒體的三羧酸 (TCA) 循環中的糖解代謝丙酮酸,並抑制氧化磷酸化,從而使癌細胞避免產生過多的活性氧 (ROS)和防止粒線體驅動細胞凋亡。One of the critical gate-keepers controlling the flow of pyruvate to lactate in the cytosol or to acetyl-CoA in mitochondria is pyruvate dehydrogenase kinase (PDK).控制細胞溶質中丙酮酸鹽向乳酸鹽流動或粒線體中乙酰輔酶A流動的關鍵守門員之一是丙酮酸脫氫酶激酶 (pyruvate dehydrogenase kinase, PDK)。PDK負調節丙酮酸脫氫酶 (pyruvate dehydrogenase, PDH),其催化丙酮酸轉化為乙酰-CoA,從而限制丙酮酸對氧化代謝的利用,同時增強腫瘤細胞優選的糖解。
龔行健榮譽研究員團隊進一步探討與前列腺癌發展有重要關聯的KDM4A及E2F1之作用機轉,提出在前列腺癌代謝過程中,組蛋白去甲基酶KDM4A為E2F1的輔激活物,並證明E2F1-KDM4A複合物在腫瘤代謝的控制中的功能作用。KDM4A及E2F1與目標基因之啟動子相關,增強E2F1染色質結合和轉錄活性,從而調節癌細胞增殖和存活所必需的轉錄型態。The pyruvate dehydrogenase kinases (PDKs) PDK1 and PDK3 are direct targets of KDM4A and E2F1 and modulate the switch between glycolytic metabolism and mitochondrial oxidation.丙酮酸脫氫酶激酶(PDK) PDK1和PDK3是KDM4A和E2F1的直接靶標,並且調節糖解代謝和粒線體氧化之間的轉換。KDM4A的下調導致丙酮酸脫氫酶(pyruvate dehydrogenase)及粒線體氧化的活性提升,造成活性氧過度的累積。The altered metabolic phenotypes can be partially rescued by ectopic expression of PDK1 and PDK3, indicating a KDM4A-dependent tumor metabolic regulation via PDK.改變的代謝表型可以透過PDK1和PDK3的異位表達部分恢復,包含經由PDK的KDM4A依賴性腫瘤代謝調控。
此研究發現KDM4A為E2F1的共激活因子,並且KDM4A-E2F複合物藉由上調PDK1和PDK3來調節代謝,從而促進氧化磷酸化向糖解代謝的轉換,為前列腺癌細胞提供增殖和存活優勢。整體研究揭示KDM4A在前列腺癌代謝過程中扮演重要角色,並且為治療之重要標的,此研究成果已發表於國際期刊Cell Reports (Wang LY, et al., Cell Reports 2016)。
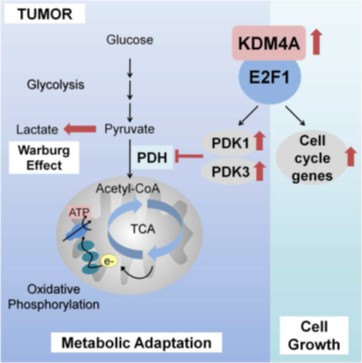
KDM4A共同激活化E2F1,以調節PDK依賴性的粒線體氧化和糖解之間的代謝轉換
Glucocorticoids經由引發miR-708的表現抑制其下游Rap1B的表現,能夠有效抑制卵巢癌細胞轉移
已知腎上腺類固醇激素的主要成分之一糖皮質激素(glucocorticoids, GCs)是對細胞的代謝、分化、增殖和存活有顯著影響的物質,本院分子與基因醫學研究所王陸海名譽研究員研究團隊與台北榮民總醫院合作,提出glucocorticoids在抑制卵巢癌轉移所扮演的嶄新角色。此研究結果揭示glucocorticoids引發微小核醣核酸miRNA-708的表現,再藉由miRNA-708抑制下游分子Rap1B的表現,阻止了經由integrin所調控的細胞移動與侵入之能力。此機制的發現,將可用以有效地抑制卵巢癌細胞的轉移。此結果已刊登在2015年1月份的國際頂尖期刊Nature Communications。
卵巢癌是女性常見癌症的第六名,全球每一年大約有225,500的婦女診斷出患有卵巢癌,其中約140,200人因而死亡,據衛生福利部統計資料,台灣地區近二十年主要婦癌(包含卵巢癌),每十萬人的發生率自4人上升至8人,其中2010年的個案數為1,245人。卵巢癌的盛行率雖然不似乳癌、口腔癌及大腸直腸癌高,但死亡率卻列為婦科腫瘤之首,發展至末期的卵巢癌患者,5年存活率不超過30%,因為幾乎沒有初期症狀,大多數患者有明顯症狀發生時多已轉移,因此,尋求預防卵巢癌轉移的新治療策略是迫切必要的課題。
本研究提出糖皮質激素glucocorticoids對卵巢癌轉移作用的嶄新調控機制。並以其中最常在臨床上使用的迪皮質醇(dexamethasone)做本研究的主要藥物進行實驗。以miRNA陣列分析具有高轉移能力的細胞株SKOV-3-I6iv細胞株以及同源的低轉移能力SKOV-3細胞株之miRNA表現情形,結果顯示miRNA-708的表現在高轉移能力的細胞株顯著偏低。進一步測試271人卵巢癌標本miR-708的表現,結果顯示晚期(III / IV期)或有轉移的原發性腫瘤miR-708的表現量,與正常卵巢組織或早期的腫瘤(I / II期)相比顯著較低。此外,在細胞模式及免疫缺陷(SCID)小鼠模式,結果皆顯示miR-708有調控腫瘤細胞侵襲及轉移的能力。於異種正位移植小鼠模式,迪皮質醇(dexamethasone)療法會誘導microRNA-708之表現,從而抑制Rap1B的表現,導致減低integrin調控細胞的焦點附著(focal adhesion)之形成,因而抑制卵巢癌細胞的遷移與侵襲,減少小鼠卵巢癌腹腔的轉移。反之,當恢復Rap1B的表現,則會反轉迪皮質醇(dexamethasone)-miR-708調控卵巢癌細胞侵襲和轉移的抑制作用。因此以Dexamethasone做腹腔注射,可以顯著的抑制種植在SCID老鼠卵巢之SKOV-3-I6iv腫瘤的腹腔轉移。臨床檢體分析顯示,卵巢癌末期呈現低表現miR-708,相較於高表現miR-708的患者,後者有較高的存活率(圖一)。圖二是miRNA-708抑制癌細胞侵襲的分子機制模式。
本研究顯示Dexamethasone或許可以被開發成抑制卵巢癌轉移的藥物。
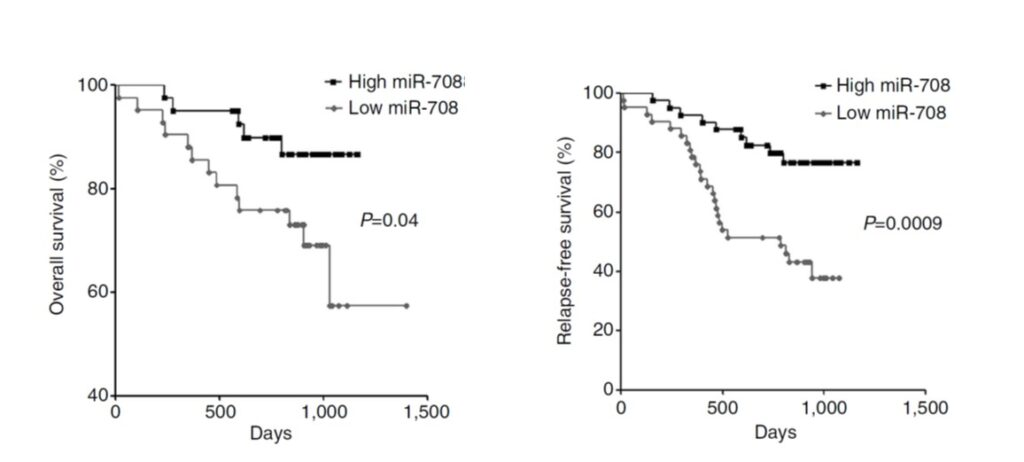
圖一 : miRNA-708表現量與卵巢癌病人存活率之相關性。
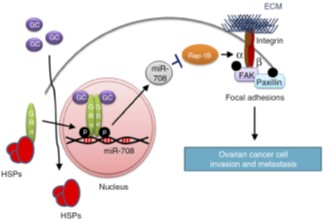
圖二 : Glucocorticoids抑制卵巢癌轉移之訊息路徑模式
腸道可以影響阿茲海默症的病程
阿茲海默症為一種最常見的神經退化性失智症,病人不只會發生記憶力逐漸衰退的現象,其認知功能和社交能力也會隨之喪失。此病好發於65歲以上之老人,在台灣80歲以上的老人每5人即有1人罹患此疾病。因為全球人口快速高齡化,阿茲海默症已儼然成為許多已開發國家重要的流行病症之一,並導致嚴重的社會與醫療的問題。很令人憂心的是,此疾病迄今並未研發出有效的藥物可以治癒或減緩阿茲海默症的病程,目前藥物只能減輕其症狀。因此,國衛院致力於研究此疾病的分子病理機制,希望將來幫助發展出新的治療策略及病人照護的注意事項。
本院分子與基因醫學研究所莊志立研究員團隊,先前與清華大學潘榮隆教授合作,共同指導博士班研究生吳師誠(目前為本院博士後研究員),利用腸道細菌餵食果蠅的幼蟲,發現這些果蠅腸道感染產生活性氧自由基後,會誘發腸道產生另外一種活性自由基(一氧化氮,NO),腸道利用這些活性自由基,將腸道感染的消息傳遞給血球細胞,然後再以這些血球細胞作為器官之間的訊號傳遞媒介,通知其他器官啟動免疫反應,揭開了腸道與其他器官之間免疫溝通的密碼。以相當於人類肝臟功能的果蠅器官脂肪體(fat body)作為實驗標的,發現當果蠅腸道活性氧自由基增加時,活化果蠅的脂肪體內NF-κB/Rel轉錄因子,果蠅的肝臟便啟動抗菌胜肽的免疫反應,引起果蠅肝臟系統性的免疫反應。此研究證實各器官的免疫反應與腸道的健康有著密切的關聯。這項研究成果已發表在2012年美國知名的細胞(Cell)期刊的系列雜誌(Cell Host & Microbe),並且獲得該期刊專文特別介紹。
腸道是神經密佈的器官,因此素有“第二大腦“之稱。因為腸道與大腦之間有密切的溝通,過去文獻的已發現一些精神疾病,例如憂鬱症或精神分裂症都與腸道菌相的失衡有關,莊研究員延續上述免疫溝通的研究發現並大膽假設:腸道細菌的失衡或許會加速阿茲海默症的病程?莊研究員率領研究團隊成員包括吳師誠博士後研究員、曹姿萱研究助理與新竹馬偕紀念醫院張國明醫師,歷經五年研究證實腸道菌相的失衡,的確會加劇阿茲海默症大腦神經退化的病程,此一前瞻創新的研究成果獲得國際高度青睞,已於2017年刊登在國際頂尖科學期刊「自然通訊」(Nature Communications)。
因為身體內器官間的緊密溝通互動,本就是動物界一種共通的生理現象,不論是高等的哺乳動物或者是昆蟲,皆靠此維繫身體生理的健康與平衡。因此,研究團隊利用果蠅阿茲海默症的動物模式,餵食腸道桿菌去破壞腸內菌相平衡。結果發現阿茲海默症的神經退化病程明顯加劇,動物壽命縮短並且行動能力變差。為了找出致病機轉,研究團隊發現腸道菌感染後,會刺激體內免疫血球細胞移動至阿茲海默症腦部,引發大腦產生大量產生促進發炎的細胞激素及氧化自由基,進而促進腦神經細胞死亡。研究團隊進一步研究,發現這些免疫血球細胞的移動能力會因腸道菌而感染提升,然後這些過動的免疫細胞會受到大腦的氧化自由基所吸引,轉移至大腦去加劇發炎反應。整體作用機制示意圖如圖一。
本研究證明腸道可以透過身體的免疫血球細胞,去控制大腦的發炎反應,進而讓阿茲海默症的神經退化加劇。雖然此觀點尚未經人體的相關研究證實,就動物器官間免疫生理平衡的共同特性,我們合理推論阿茲海默症的病人應避免腸道感染以免加劇病情。另外,我們雖然目前看到的是腸道對大腦的負面影響,若純粹只以兩者溝通的角色來思考,我們的發現似乎也暗示著腸道與大腦之間的免疫溝通,是一種控制大腦發炎以及神經退化性的重要機制。因此,我們並不排除腸道也可能正面去影響大腦的健康。此假設若未來經研究證實,我們就可以利用藥物或飲食運動調控腸道功能,去間接改善大腦的健康。未來治療阿茲海默症,或許就不需要依靠藥物穿透腦血屏障進入大腦,只要透過改變腸道與大腦的免疫溝通,就能減緩阿茲海默症的病程。此研究的發現或許可幫助未來在研發阿茲海默症的治療策略上,提供阿茲海默症的新方向。
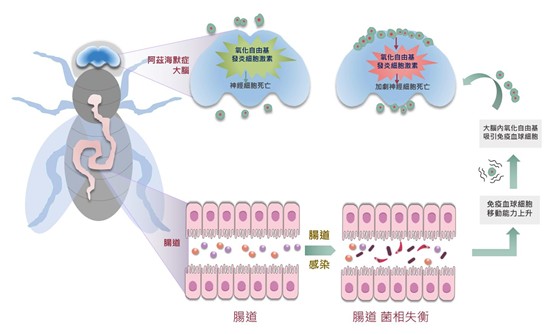 圖一、腸道菌感染後,會刺激體內免疫血球細胞移動至阿茲海默症腦部,引發大腦產生大量產生促進發炎的細胞激素及氧化自由基,進而促進腦神經細胞死亡。
圖一、腸道菌感染後,會刺激體內免疫血球細胞移動至阿茲海默症腦部,引發大腦產生大量產生促進發炎的細胞激素及氧化自由基,進而促進腦神經細胞死亡。